This is the AR & FOXA1 notebook executed end to end, with its figures and result tables. The previous pages explain the concepts; this one shows the actual run. Source:
examples/ar_foxa1_lncap/.
AR & FOXA1 in LNCaP (±DHT)
How the androgen receptor (AR) cistrome depends on the pioneer factor FOXA1 after androgen (DHT) stimulation in LNCaP cells.
Data are ChIP-Atlas (hg38) peaks + bigwigs — run ./download_data.sh first.
LNCaP, 0h vs 4h DHT, for FOXA1, AR, and ATAC-seq (two ATAC replicates / condition).
Note — FOXA1 4h peaks use
SRX23002841.05.bed(matching the FOXA1 4h bigwig). The reference paths in the next cell point at the lackgrp cluster; edit them for your environment.
0. Setup — paths & helpers
import os, time
from contextlib import contextmanager
import numpy as np
import matplotlib.pyplot as plt
from tqdm import tqdm
import genomeblocks as gb
from genomeblocks import Loci, Genes, Atlas, make_genome, tmm
from genomeblocks.signal_draw import plot_heatmap
from genomeblocks.motifs import scan_motifs_matrix, bootstrap_enrichment
from genomeblocks import browser
@contextmanager
def timer(label):
"""Print a step's wall-clock time."""
t0 = time.perf_counter()
print(f"\u25b6 {label} ...")
try:
yield
finally:
print(f"\u2713 {label} \u2014 {time.perf_counter() - t0:.1f}s")
# --- downloaded data (see download_data.sh) ---
DATA = "data"
DATA
SRX = {
"FOXA1_0h": "SRX23002839", "FOXA1_4h": "SRX23002841",
"AR_0h": "SRX23002834", "AR_4h": "SRX23002836",
"ATAC_0h_r1": "SRX23002894", "ATAC_0h_r2": "SRX23002895",
"ATAC_4h_r1": "SRX23002898", "ATAC_4h_r2": "SRX23002899",
}
PEAK = {k: f"{DATA}/{s}.05.bed" for k, s in SRX.items()}
BW = {k: f"{DATA}/{s}.bw" for k, s in SRX.items()}
# --- hg38 references (edit for your environment) ---
GENOME_FA = "/groups/lackgrp/genome_annotations/hg38/hg38.fa"
GTF = "/groups/lackgrp/genome_annotations/hg38/gencode.v49.annotation_protein_coding.gtf"
MOTIF_DB = "/groups/lackgrp/databases/motifs/motif-db/H14CORE_jaspar_format_lightmotif.txt"
CHROMSIZES = "/groups/lackgrp/genome_annotations/hg38/hg38_chr.chrom.sizes"
GIGGLE_META = "/groups/lackgrp/databases/giggle/giggle_hg38/hg38_tfs_meta.tsv"
GIGGLE_DIR = "/groups/lackgrp/databases/giggle/giggle_hg38/ChIP-Atlas-ALL"
1. Load peaks
with timer("load peak files"):
peaks = {k: Loci.make(v) for k, v in PEAK.items()}
for k, v in peaks.items():
print(f"{k:12} {len(v):>8,} peaks")
▶ load peak files ...
✓ load peak files — 0.6s
FOXA1_0h 8,657 peaks
FOXA1_4h 10,870 peaks
AR_0h 910 peaks
AR_4h 3,645 peaks
ATAC_0h_r1 53,671 peaks
ATAC_0h_r2 62,741 peaks
ATAC_4h_r1 47,780 peaks
ATAC_4h_r2 51,196 peaks
2. Accessible chromatin — union of ATAC peaks
Concatenate the four ATAC peak sets (both conditions, both reps) and merge()
overlapping intervals into one accessible-region set.
with timer("ATAC union (accessible regions)"):
accessible = (peaks["ATAC_0h_r1"] + peaks["ATAC_0h_r2"]
+ peaks["ATAC_4h_r1"] + peaks["ATAC_4h_r2"]).sort().merge()
print(f"accessible regions (merged): {len(accessible):,}")
▶ ATAC union (accessible regions) ...
✓ ATAC union (accessible regions) — 0.7s
accessible regions (merged): 72,470
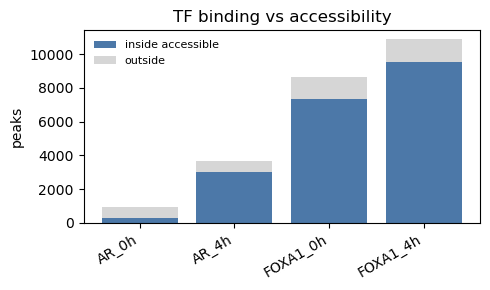
3. AR / FOXA1 binding inside vs outside accessible chromatin
rows = []
for name in ["AR_0h", "AR_4h", "FOXA1_0h", "FOXA1_4h"]:
s = peaks[name]
inside = len(s & accessible) # peaks overlapping accessible
total = len(s)
rows.append((name, inside, total - inside))
print(f"{name:10} {100 * inside / total:5.1f}% inside accessible ({inside:,}/{total:,})")
names = [r[0] for r in rows]
ins = np.array([r[1] for r in rows])
outs = np.array([r[2] for r in rows])
fig, ax = plt.subplots(figsize=(5, 3))
ax.bar(names, ins, label="inside accessible", color="#4c78a8")
ax.bar(names, outs, bottom=ins, label="outside", color="#d6d6d6")
ax.set_ylabel("peaks"); ax.legend(frameon=False, fontsize=8)
ax.set_title("TF binding vs accessibility")
plt.xticks(rotation=30, ha="right"); plt.tight_layout()
AR_0h 31.2% inside accessible (284/910)
AR_4h 83.0% inside accessible (3,027/3,645)
FOXA1_0h 85.0% inside accessible (7,362/8,657)
FOXA1_4h 88.0% inside accessible (9,561/10,870)
4. Keep only accessible TF peaks
with timer("filter TF peaks to accessible"):
F0 = peaks["FOXA1_0h"] & accessible
F4 = peaks["FOXA1_4h"] & accessible
A4 = peaks["AR_4h"] & accessible
print(f"accessible FOXA1 0h={len(F0):,} FOXA1 4h={len(F4):,} AR 4h={len(A4):,}")
▶ filter TF peaks to accessible ...
✓ filter TF peaks to accessible — 0.0s
accessible FOXA1 0h=7,362 FOXA1 4h=9,561 AR 4h=3,027
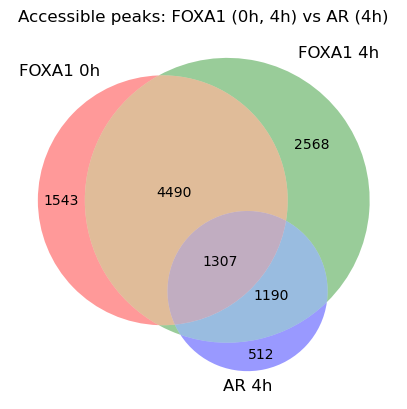
5. Venn — FOXA1 (0h, 4h) vs AR (4h)
To venn genomic intervals we merge all peaks into a shared region universe, then label each region by which input set overlaps it. From this:
- AR+F = AR 4h peaks that overlap FOXA1 (0h or 4h) — FOXA1-dependent AR
- AR−F = AR 4h peaks that overlap no FOXA1 peak — FOXA1-independent AR
from matplotlib_venn import venn3
def venn_id_sets(sets):
"""Merge all peaks into a region universe; return one set of region-ids per
input set (the ids it overlaps) so matplotlib_venn can count the regions."""
universe = Loci([l for s in sets for l in s]).sort().merge()
id_sets = [set() for _ in sets]
for i, reg in enumerate(tqdm(universe, desc="venn membership")):
for si, s in enumerate(sets):
if any(True for _ in s.cgr.overlap(reg.chrom, reg.start, reg.end)):
id_sets[si].add(i)
return id_sets
with timer("venn3 membership"):
ids = venn_id_sets([F0, F4, A4])
fig, ax = plt.subplots(figsize=(5, 5))
venn3(ids, set_labels=["FOXA1 0h", "FOXA1 4h", "AR 4h"], ax=ax)
ax.set_title("Accessible peaks: FOXA1 (0h, 4h) vs AR (4h)")
▶ venn3 membership ...
venn membership: 100%|██████████| 11624/11624 [00:00<00:00, 238333.00it/s]
✓ venn3 membership — 0.1s
Text(0.5, 1.0, 'Accessible peaks: FOXA1 (0h, 4h) vs AR (4h)')
with timer("define AR+F / AR-F"):
F_any = (F0 + F4).sort().merge() # FOXA1-bound at either timepoint
ARpF = A4 & F_any # AR 4h that IS FOXA1-bound
ARmF = A4 - F_any # AR 4h that is NOT FOXA1-bound
print(f"AR+F (FOXA1-dependent): {len(ARpF):,} AR-F (FOXA1-independent): {len(ARmF):,}")
▶ define AR+F / AR-F ...
✓ define AR+F / AR-F — 0.0s
AR+F (FOXA1-dependent): 2,515 AR-F (FOXA1-independent): 512
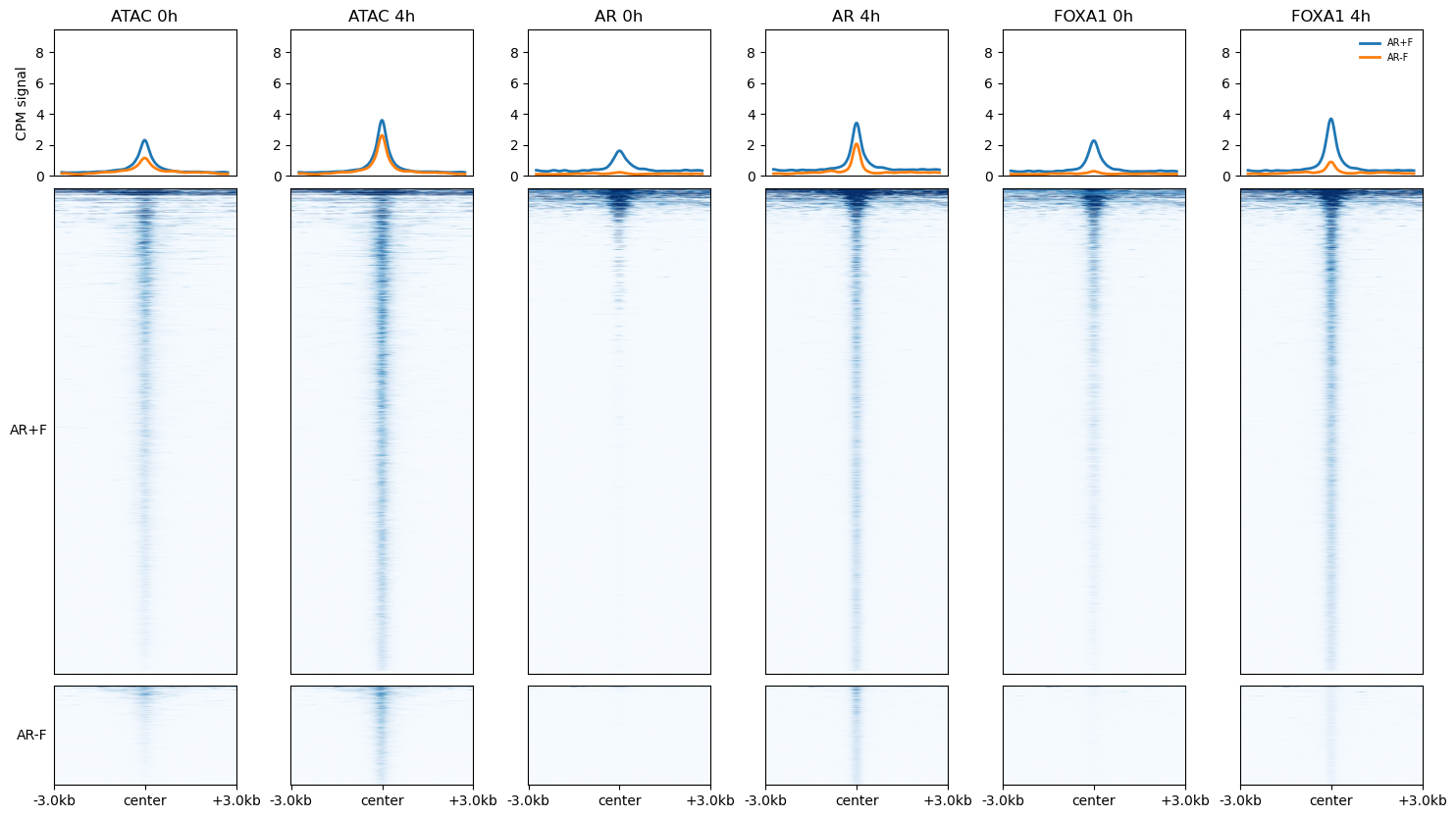
6. Signal heatmaps across conditions
Extract signal for AR+F and AR−F over all 8 bigwigs, average the two ATAC replicates per condition (the same averaging the browser does), and draw a grouped heatmap: ATAC 0h, ATAC 4h, AR 0h, AR 4h, FOXA1 0h, FOXA1 4h.
regions = ARpF + ARmF
groups = {"AR+F": ARpF, "AR-F": ARmF}
bw_order = ["ATAC_0h_r1", "ATAC_0h_r2", "ATAC_4h_r1", "ATAC_4h_r2",
"AR_0h", "AR_4h", "FOXA1_0h", "FOXA1_4h"]
bigwigs = [BW[k] for k in bw_order]
with timer("extract signal cube"):
S = np.nan_to_num(regions.signal(bigwigs, n_bins=200, flank=2000, workers=4))
#S = tmm(np.nan_to_num(S)) # per-track normalization
# group ATAC replicates by averaging their columns -> 6 conditions
S6 = np.stack([
S[:, [0, 1], :].mean(1), # ATAC 0h (rep1+rep2)
S[:, [2, 3], :].mean(1), # ATAC 4h (rep1+rep2)
S[:, 4, :], S[:, 5, :], # AR 0h, AR 4h
S[:, 6, :], S[:, 7, :], # FOXA1 0h, FOXA1 4h
], axis=1)
samples = ["ATAC 0h", "ATAC 4h", "AR 0h", "AR 4h", "FOXA1 0h", "FOXA1 4h"]
vmax = float(np.percentile(S6, 99))
with timer("draw heatmap"):
fig = plot_heatmap(regions, S6, groups=groups, sets=["AR+F", "AR-F"],
samples=samples, vmax=vmax, ymax=vmax)
▶ extract signal cube ...
[INFO] Extracting 8 bigwigs for 3027 loci into 200 bins (span=False, agg='mean', backend='pybigtools', exact=True, workers=4).
chunks: 100%|██████████| 4/4 [00:03<00:00, 1.16it/s]
✓ extract signal cube — 3.6s
▶ draw heatmap ...
✓ draw heatmap — 0.1s
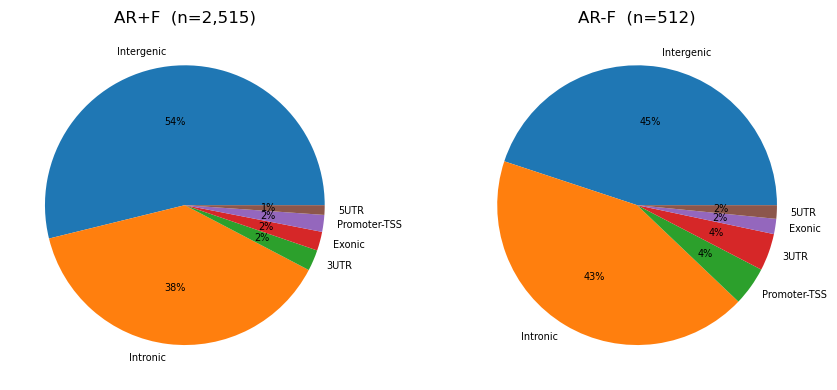
7. Genomic annotation of AR+F vs AR−F
with timer("load genes (GTF)"):
genes = Genes.make(GTF)
with timer("annotate AR+F / AR-F"):
annot_pf = genes.annotations(ARpF)
annot_mf = genes.annotations(ARmF)
fig, axes = plt.subplots(1, 2, figsize=(9, 4))
for ax, df, title in [(axes[0], annot_pf, "AR+F"), (axes[1], annot_mf, "AR-F")]:
vc = df["annotation"].value_counts()
ax.pie(vc.values, labels=vc.index, autopct="%1.0f%%", textprops={"fontsize": 7})
ax.set_title(f"{title} (n={len(df):,})")
plt.tight_layout()
▶ load genes (GTF) ...
[INFO] Parsing GTF/GFF file 🧩: 6731674it [01:05, 103025.37it/s]
[INFO] Unmapped feature types: start_codon, Selenocysteine, stop_codon
✓ load genes (GTF) — 65.5s
▶ annotate AR+F / AR-F ...
✓ annotate AR+F / AR-F — 16.5s
8. Motif enrichment — AR+F vs AR−F
Scan JASPAR motifs over AR+F, AR−F, and a subsample of the accessible (ATAC)
pool as background, then bootstrap the log-fold-change. Sorting by LFC
(= LFC_AR+F − LFC_AR−F) gives motifs preferential to each set.
with timer("load genome FASTA"):
genome = make_genome(GENOME_FA)
# background pool = subsample of accessible chromatin (keeps scanning cheap)
pool = accessible
with timer("scan motifs (AR+F, AR-F, pool)"):
mat_pf = scan_motifs_matrix(ARpF, genome, MOTIF_DB, r=250, workers=8)
mat_mf = scan_motifs_matrix(ARmF, genome, MOTIF_DB, r=250, workers=8)
mat_pool = scan_motifs_matrix(pool, genome, MOTIF_DB, r=250, workers=8)
with timer("bootstrap enrichment"):
enr = bootstrap_enrichment({"AR+F": mat_pf, "AR-F": mat_mf},
ref=mat_pool, boot=100, sample=500, seed=0)
show = ["Factor", "LFC", "LFC_AR+F", "LFC_AR-F"]
ranked = enr.sort_values("LFC", ascending=False)
print("Top motifs in AR+F (FOXA1-dependent):")
display(ranked.head(10)[show])
print("Top motifs in AR-F (FOXA1-independent):")
display(ranked.tail(10)[show].iloc[::-1])
▶ load genome FASTA ...
✓ load genome FASTA — 20.4s
▶ scan motifs (AR+F, AR-F, pool) ...
[motifs]: 100%|██████████| 33/33 [00:01<00:00, 29.99it/s]
[motifs]: 100%|██████████| 33/33 [00:00<00:00, 118.08it/s]
[motifs]: 100%|██████████| 33/33 [00:30<00:00, 1.08it/s]
✓ scan motifs (AR+F, AR-F, pool) — 55.5s
▶ bootstrap enrichment ...
100%|██████████| 100/100 [00:00<00:00, 121.79it/s]
100%|██████████| 100/100 [00:00<00:00, 140.44it/s]
100%|██████████| 100/100 [00:00<00:00, 261.65it/s]
✓ bootstrap enrichment — 2.4s
Top motifs in AR+F (FOXA1-dependent):
| Factor | LFC | LFC_AR+F | LFC_AR-F | |
|---|---|---|---|---|
| 1439 | ZN671.H14CORE.0.P.C | 0.200354 | 0.189548 | -0.010806 |
| 1098 | TSH2.H14CORE.0.SG.A | 0.149624 | 0.145360 | -0.004264 |
| 240 | FOXA2.H14CORE.0.PSM.A | 0.123507 | 0.073603 | -0.049904 |
| 242 | FOXA3.H14CORE.0.PS.A | 0.106075 | 0.077160 | -0.028915 |
| 265 | FOXL2.H14CORE.0.PSM.A | 0.094850 | 0.072495 | -0.022355 |
| 266 | FOXM1.H14CORE.0.P.B | 0.091278 | 0.073294 | -0.017984 |
| 271 | FOXP1.H14CORE.0.PS.A | 0.088453 | 0.072825 | -0.015627 |
| 262 | FOXK1.H14CORE.0.PS.A | 0.087973 | 0.059407 | -0.028566 |
| 238 | FOXA1.H14CORE.0.P.B | 0.079758 | 0.051366 | -0.028392 |
| 268 | FOXO3.H14CORE.0.PS.A | 0.068372 | 0.043914 | -0.024458 |
Top motifs in AR-F (FOXA1-independent):
| Factor | LFC | LFC_AR+F | LFC_AR-F | |
|---|---|---|---|---|
| 7 | ANDR.H14CORE.0.P.B | -0.153016 | 0.078426 | 0.231442 |
| 836 | PRGR.H14CORE.0.P.B | -0.090640 | 0.068276 | 0.158916 |
| 293 | GCR.H14CORE.0.PS.A | -0.085943 | 0.034013 | 0.119956 |
| 491 | KLF16.H14CORE.1.P.B | -0.082473 | -0.244051 | -0.161577 |
| 553 | MAZ.H14CORE.1.P.B | -0.072905 | -0.176444 | -0.103539 |
| 1559 | ZNF48.H14CORE.0.PSG.A | -0.071781 | -0.042426 | 0.029356 |
| 1122 | VEZF1.H14CORE.1.P.B | -0.070181 | -0.129904 | -0.059723 |
| 506 | KMT2A.H14CORE.0.P.B | -0.057221 | -0.300223 | -0.243002 |
| 557 | MCR.H14CORE.0.S.B | -0.057109 | 0.027812 | 0.084921 |
| 1342 | ZN467.H14CORE.0.P.C | -0.056477 | -0.088766 | -0.032288 |
9. ChIP-Atlas (atlas) differential enrichment
Build a 1 kb bin × track index over the ChIP-Atlas hg38 collection, then use each region set as the other’s reference to find TF tracks differentially enriched between AR+F and AR−F.
Building the index over the full ChIP-Atlas is heavy (minutes, several GB RAM). Pickle
atlasto reuse it across sessions.
with timer("build ChIP-Atlas index (1kb)"):
atlas = Atlas.make(
GIGGLE_DIR, chromsizes=CHROMSIZES,
meta=GIGGLE_META,
name_pattern=r"([^.]+)", # SRX23002840.20.bed.gz -> SRX23002840
meta_columns=["id", "antigen", "class", "cell_line"], # meta TSV is header-less
meta_id_col="id",
)
with timer("atlas search AR+F vs AR-F"):
enr_pf = atlas.search(ARpF, ref=ARmF) # enriched in AR+F over AR-F
enr_mf = atlas.search(ARmF, ref=ARpF) # enriched in AR-F over AR+F
cols = ["name", "antigen", "class", "cell_line", "overlaps", "log2_odds", "giggle_score"]
print("TF tracks enriched in AR+F (FOXA1-dependent):")
display(enr_pf.head(15)[cols])
print("TF tracks enriched in AR-F (FOXA1-independent):")
display(enr_mf.head(15)[cols])
▶ build ChIP-Atlas index (1kb) ...
[atlas index]: 100%|██████████| 33368/33368 [00:19<00:00, 1726.86it/s]
✓ build ChIP-Atlas index (1kb) — 60.5s
▶ atlas search AR+F vs AR-F ...
✓ atlas search AR+F vs AR-F — 4.6s
TF tracks enriched in AR+F (FOXA1-dependent):
| name | antigen | class | cell_line | overlaps | log2_odds | giggle_score | |
|---|---|---|---|---|---|---|---|
| 0 | SRX23002840 | FOXA1 | Prostate | LNCAP | 1329 | 7.519583 | 964.923631 |
| 1 | SRX23002841 | FOXA1 | Prostate | LNCAP | 1089 | 7.056433 | 700.212091 |
| 2 | SRX18285237 | FOXA1 | Prostate | LNCAP | 1847 | 4.287255 | 618.551089 |
| 3 | SRX14353424 | FOXA1 | Prostate | LNCAP | 1990 | 4.058525 | 604.652460 |
| 4 | SRX18285235 | FOXA1 | Prostate | LNCAP | 1722 | 4.333244 | 580.711304 |
| 5 | SRX062360 | FOXA1 | Prostate | LNCAP | 1564 | 4.546911 | 564.964852 |
| 6 | SRX14353423 | FOXA1 | Prostate | LNCAP | 1756 | 4.155838 | 548.736062 |
| 7 | SRX5577144 | Epitope tags | Prostate | LNCAP | 1779 | 4.090175 | 540.034633 |
| 8 | SRX1885188 | FOXA1 | Prostate | LNCAP | 2044 | 3.775184 | 533.691700 |
| 9 | SRX18285236 | FOXA1 | Prostate | LNCAP | 1470 | 4.498439 | 515.002750 |
| 10 | SRX1885186 | FOXA1 | Prostate | LNCAP | 1936 | 3.774294 | 503.551086 |
| 11 | SRX1885187 | FOXA1 | Prostate | LNCAP | 1878 | 3.818223 | 499.236335 |
| 12 | SRX1885185 | FOXA1 | Prostate | LNCAP | 1972 | 3.724534 | 499.063986 |
| 13 | SRX1212235 | FOXA1 | Prostate | LNCAP | 1936 | 3.749926 | 496.615360 |
| 14 | SRX6878606 | FOXA1 | Prostate | LNCAP | 1877 | 3.763680 | 484.060928 |
TF tracks enriched in AR-F (FOXA1-independent):
| name | antigen | class | cell_line | overlaps | log2_odds | giggle_score | |
|---|---|---|---|---|---|---|---|
| 0 | SRX3070471 | NR3C1 | Breast | MCF 10A | 151 | 1.985641 | 57.274594 |
| 1 | SRX3070475 | NR3C1 | Breast | MCF 10A | 120 | 2.021899 | 49.016228 |
| 2 | SRX21439743 | ESR1 | Prostate | LNCAP | 452 | 1.518987 | 46.612336 |
| 3 | SRX3070473 | NR3C1 | Breast | MCF 10A | 120 | 1.969408 | 45.866518 |
| 4 | SRX306516 | AR | Prostate | DU 145 | 161 | 1.690385 | 39.808143 |
| 5 | SRX19970679 | AR | Prostate | PC-346C | 167 | 1.537133 | 31.894465 |
| 6 | SRX8520802 | NR3C1 | Breast | HCC1187 | 173 | 1.506006 | 31.057426 |
| 7 | SRX21439744 | ESR1 | Prostate | LNCAP | 336 | 1.292770 | 30.240255 |
| 8 | SRX3070469 | NR3C1 | Breast | MCF 10A | 53 | 2.255766 | 30.173945 |
| 9 | SRX21439742 | ESR1 | Prostate | LNCAP | 295 | 1.284194 | 28.358706 |
| 10 | SRX083218 | AR | Prostate | LNCAP | 236 | 1.337458 | 28.128496 |
| 11 | SRX1067071 | AR | Prostate | LHSAR | 319 | 1.253582 | 27.290928 |
| 12 | SRX1067070 | AR | Prostate | LHSAR | 340 | 1.245091 | 27.235381 |
| 13 | SRX083219 | AR | Prostate | LNCAP | 233 | 1.311355 | 26.426950 |
| 14 | SRX3630817 | NR3C1 | Uterus | Ishikawa | 53 | 2.092061 | 25.168441 |
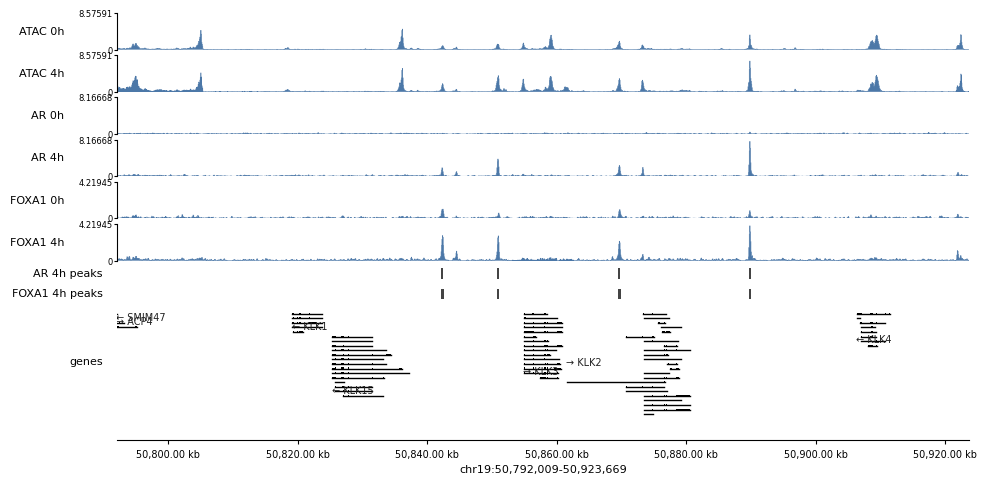
10. Browser view — chr19:50,792,009-50,923,669
All six conditions as signal tracks (ATAC replicates passed as a list of bigwigs are averaged into one track), their peak calls, and gene models.
region = "chr19:50,792,009-50,923,669"
tracks = {
"ATAC 0h": [BW["ATAC_0h_r1"], BW["ATAC_0h_r2"]], # list -> averaged
"ATAC 4h": [BW["ATAC_4h_r1"], BW["ATAC_4h_r2"]],
"AR 0h": BW["AR_0h"],
"AR 4h": BW["AR_4h"],
"FOXA1 0h": BW["FOXA1_0h"],
"FOXA1 4h": BW["FOXA1_4h"],
"AR 4h peaks": PEAK["AR_4h"],
"FOXA1 4h peaks": PEAK["FOXA1_4h"],
"genes": genes,
}
# share the y-axis within each assay so the 0h -> 4h gain is honest
with timer("browser render"):
fig, axes = browser(region, tracks, bw_n_bins=2000, figsize=(11, None),
bw_share=[["ATAC 0h", "ATAC 4h"],
["AR 0h", "AR 4h"],
["FOXA1 0h", "FOXA1 4h"]])
▶ browser render ...
✓ browser render — 1.3s